Fluorinated Analogues to the Pentuloses of the Pentose Phosphate Pathway
Fluorinated Analogues to the Pentuloses of the Pentose Phosphate Pathway
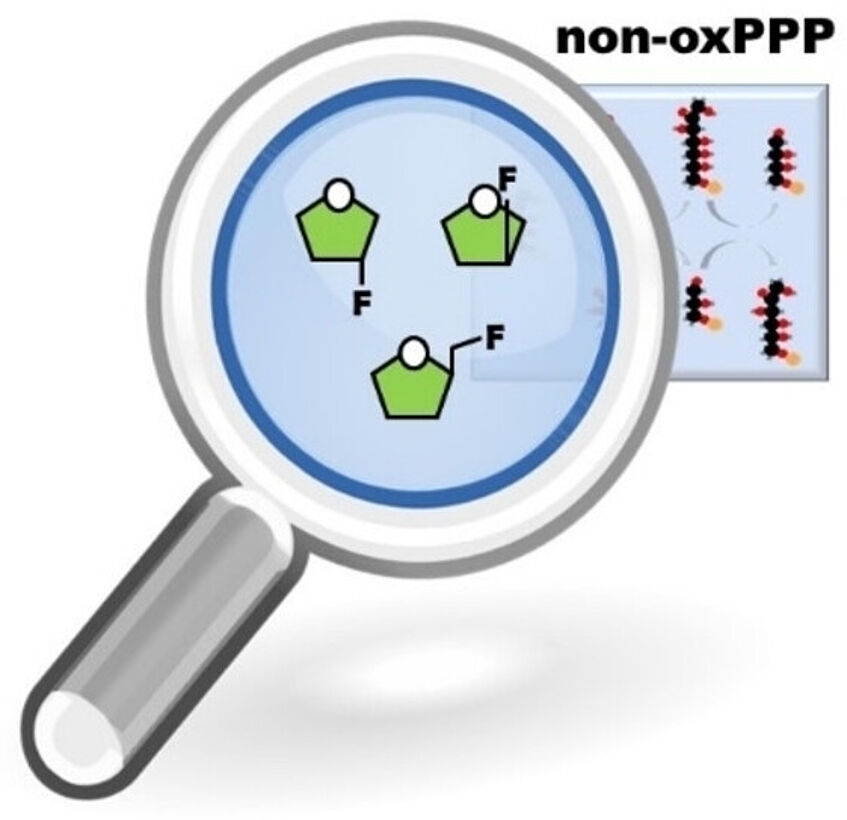
Fluorinated carbohydrates are valuable tools for enzymological studies due to their increased metabolic stability compared to their non-fluorinated analogues. Replacing different hydroxyl groups within the same monosaccharide by fluorine allows to influence a wide range of sugar–receptor interactions and enzymatic transformations. In the past, this principle was frequently used to study the metabolism of highly abundant carbohydrates, while the metabolic fate of rare sugars is still poorly studied. Rare sugars, however, are key intermediates of many metabolic routes, such as the pentose phosphate pathway (PPP). Here we present the design and purely chemical synthesis of a set of three deoxyfluorinated analogues of the rare sugars d-xylulose and d-ribulose: 1-deoxy-1-fluoro-d-ribulose (1DFRu), 3-deoxy-3-fluoro-d-ribulose (3DFRu) and 3-deoxy-3-fluoro-d-xylulose (3DFXu). Together with a designed set of potential late-stage radio-fluorination precursors, they have the potential to become useful tools for studies on the complex equilibria of the non-oxidative PPP (Eur. J. Org. Chem. 2023, 26, e202300339).
MAPkinases regulate secondary metabolism, sexual development and light dependent cellulase regulation in Trichoderma reesei
The filamentous fungus Trichoderma reesei is a prolific producer of plant cell wall degrading enzymes, which are regulated in response to diverse environmental signals for optimal adaptation, but also produces a wide array of secondary metabolites. Available carbon source and light are the strongest cues currently known to impact secreted enzyme levels and an interplay with regulation of secondary metabolism became increasingly obvious in recent years. While cellulase regulation is already known to be modulated by different mitogen activated protein kinase (MAPK) pathways, the relevance of the light signal, which is transmitted by this pathway in other fungi as well, is still unknown in T. reesei as are interconnections to secondary metabolism and chemical communication under mating conditions. Here we show that MAPkinases differentially influence cellulase regulation in light and darkness and that the Hog1 homologue TMK3, but not TMK1 or TMK2 are required for the chemotropic response to glucose in T. reesei. Additionally, MAPkinases regulate production of specific secondary metabolites including trichodimerol and bisorbibutenolid, a bioactive compound with cytostatic effect on cancer cells and deterrent effect on larvae, under conditions facilitating mating, which reflects a defect in chemical communication. Strains lacking either of the MAPkinases become female sterile, indicating the conservation of the role of MAPkinases in sexual fertility also in T. reesei. In summary, our findings substantiate the previously detected interconnection of cellulase regulation with regulation of secondary metabolism as well as the involvement of MAPkinases in light dependent gene regulation of cellulase and secondary metabolite genes in fungi (Sci. Rep. 2023, 13, 1912).
Direct enantioselective α-amination of amides guided by DFT prediction of E/Z selectivity in a sulfonium intermediate
Direct enantioselective α-amination of amides guided by DFT prediction of E/Z selectivity in a sulfonium intermediate
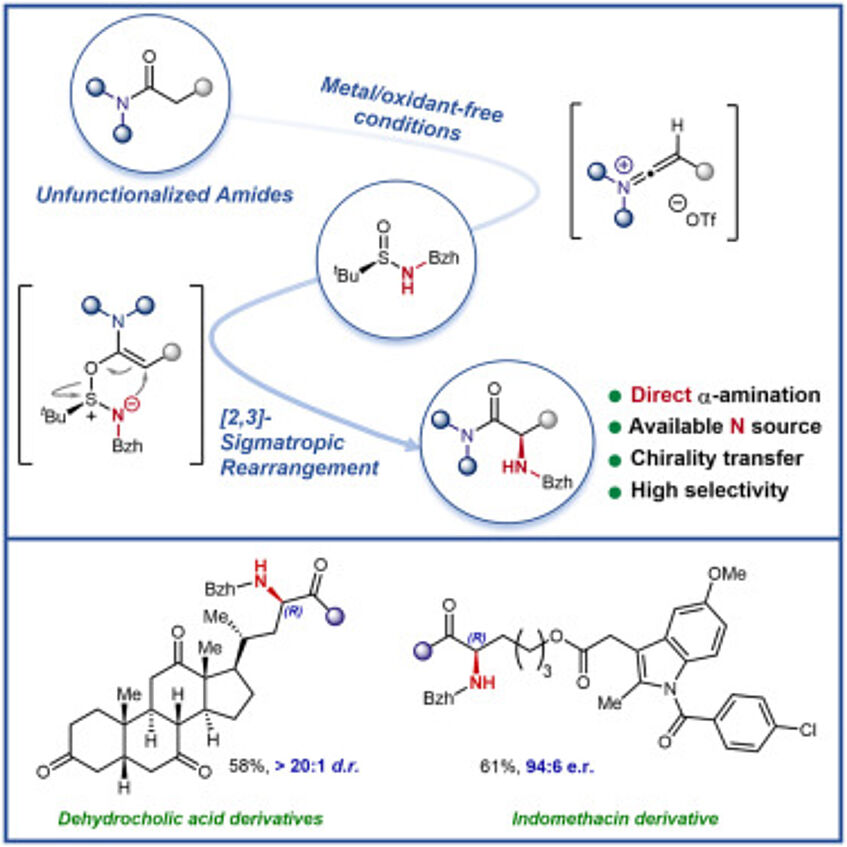
α-Amino acid derivatives are among the most important families of natural products and are essential molecules in many scientific areas. They are continuously employed in the elaboration of peptides and protein synthesis and can serve as chiral catalysts or chiral-pool starting materials in asymmetric synthesis. We report here a method for the preparation of enantioenriched natural and non-natural α-amino acid derivatives by the straightforward enantioselective amination of unfunctionalized amides with readily available chiral sulfinamides in one step. The transformation shows excellent selectivity on substrates bearing a large variety of (potentially competing) functional groups and is entirely reagent controlled. We believe that this procedure will help streamline access to non-natural α-amino acid derivatives (Chem 2023, 9, 1538–1548).
Biomimetic Cationic Cyclopropanation Enables an Efficient Chemoenzymatic Synthesis of 6,8-Cycloeudesmanes
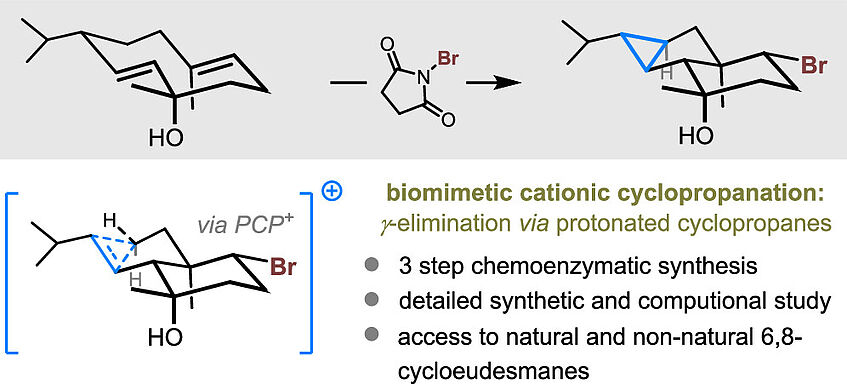
Cationic cyclopropanation involves the γ-elimination at carbocations to form a new σ-C–C bond through proton loss. While exceedingly rare in bulk solution, it is recognized as one of the main biosynthetic cyclopropanation pathways. Despite the rich history of bioinspired synthetic chemistry, cationic cyclopropanation has not been appropriated for the synthetic toolbox, likely due to the preference of carbocations to undergo competing E1 β-elimination pathways. Here, we present an in-depth synthetic and computational study of cationic cyclopropanation, focusing on the 6,8-cycloeudesmanes as a platform for this investigation. We were able to apply biomimetic cationic cyclopropanation to the synthesis of several 6,8-cycloeudesmanes and non-natural analogues─in doing so, we showcase the power of this transformation in the preparation of complex cyclopropanes (J. Am. Chem. Soc. 2023, 145, 5855–5863).
Customising excitation properties of polycyclic aromatic hydrocarbons by rational positional heteroatom doping: the peri-xanthenoxanthene (PXX) case
Customising excitation properties of polycyclic aromatic hydrocarbons by rational positional heteroatom doping: the peri-xanthenoxanthene (PXX) case
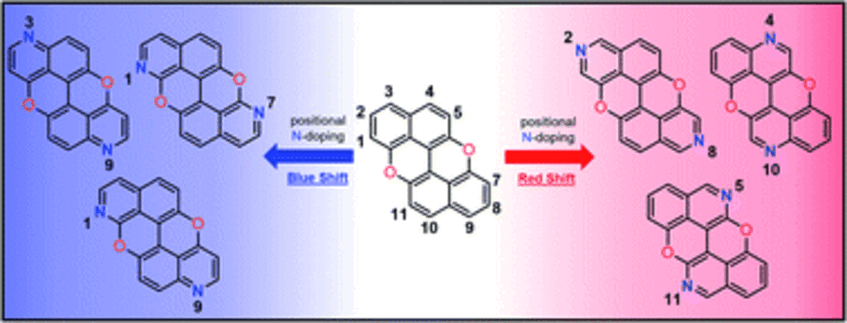
In this paper we tackle the challenge of gaining control of the photophysical properties of PAHs through a site-specific N-doping within the structural aromatic framework. By developing a simple predictive tool that identifies C(sp<small>2</small>)-positions that if substituted with a heteroatom would tailor the changes in the absorption and emission spectral envelopes, we predict optimal substitutional patterns for the model peri-xanthenoxanthene (PXX) PAH. Specifically, TDDFT calculations of the electron density difference between the S<small>1</small> excited state and S<small>0</small> ground state of PXX allowed us to identify the subtleties in the role of sites i.e., electron donating or withdrawing character on excitation. The replacement of two C(sp<small>2</small>)-atoms with two N-atoms, in either electron donating or withdrawing positions, shifts the electronic transitions either to low or high energy, respectively. This consequently shifts the PXX absorption spectral envelop bathochromically or hypsochromically, as demonstrated by steady-state absorption spectroscopic measurements. Within the series of synthesised N-doped PXX, we tune the optical band gap within an interval of ∼0.4 eV, in full agreement with the theoretical predictions. Relatedly, measurements show the more blueshifted the absorption/emission energies, the greater the fluorescence quantum yield value (from ∼45% to ∼75%). On the other hand, electrochemical investigations suggested that the N-pattern has a limited influence on the redox properties. Lastly, depending on the N-pattern, different supramolecular organisations could be obtained at the solid-state, with the 1,7-pattern PXX molecule forming multi-layered, graphene-like, supramolecular sheets through a combination of weak H-bonding and π–π stacking interactions. Supramolecular striped patterned sheets could also be formed with the 3,9- and 4,10-congeners when co-crystallized with a halogen-bond donor molecule (Chem. Sci. 2022, 13, 6335–6347).
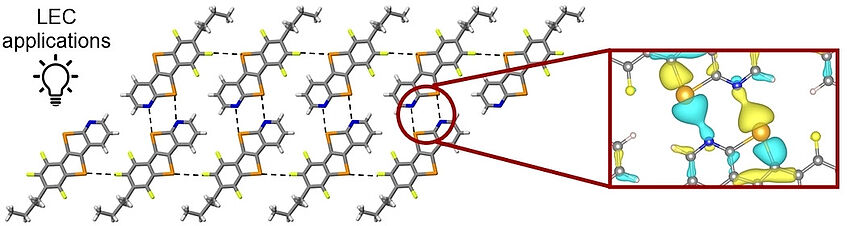
Supramolecular Chalcogen-Bonded Semiconducting Nanoribbons at Work in Lighting Devices
This work describes the design and synthesis of a π-conjugated telluro[3,2-β][1]-tellurophene-based synthon that, embodying pyridyl and haloaryl chalcogen-bonding acceptors, self-assembles into nanoribbons through chalcogen bonds. The ribbons π-stack in a multi-layered architecture both in single crystals and thin films. Theoretical studies of the electronic states of chalcogen-bonded material showed the presence of a local charge density between Te and N atoms. OTFT-based charge transport measurements showed hole-transport properties for this material. Its integration as a p-type semiconductor in multi-layered CuI-based light-emitting electrochemical cells (LECs) led to a 10-fold increase in stability (38 h vs. 3 h) compared to single-layered devices. Finally, using the reference tellurotellurophene congener bearing a C−H group instead of the pyridyl N atom, a herringbone solid-state assembly is formed without charge transport features, resulting in LECs with poor stabilities (<1 h) (Angew. Chem. Int. Ed. 2022, 61, e202202137).
peri-Acenoacene Ribbons with Zigzag BN-Doped Peripheries
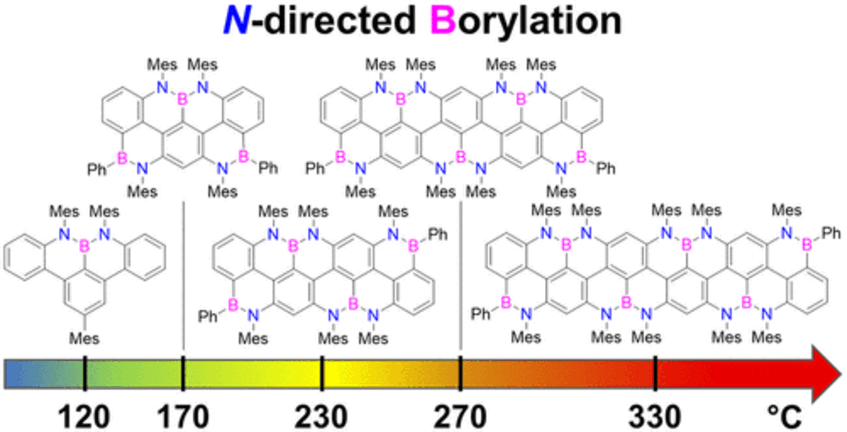
peri-Acenoacene Ribbons with Zigzag BN-Doped Peripheries
Here, we report the synthesis of BN-doped graphenoid nanoribbons, in which peripheral carbon atoms at the zigzag edges have been selectively replaced by boron and nitrogen atoms as BN and NBN motifs. This includes high-yielding ring closure key steps that, through N-directed borylation reaction using solely BBr3, allow the planarization of meta-oligoarylenyl precursors, through the formation of B–N and B–C bonds, to give ter-, quater-, quinque-, and sexi-arylenyl nanoribbons. X-ray single-crystal diffraction studies confirmed the formation of the BN and NBN motifs and the zigzag-edged topology of the regularly doped ribbons. Steady-state absorption and emission investigations at room temperature showed a systematic bathochromic shift of the UV–vis absorption and emission envelopes upon elongation of the oligoarylenyl backbone, with the nanoribbon emission featuring a TADF component. All derivatives displayed phosphorescence at 77 K. Electrochemical studies showed that the π-extension of the peri-acenoacene framework provokes a lowering of the first oxidative event (from 0.83 to 0.40 V), making these nanoribbons optimal candidates to engineer p-type organic semiconductors (J. Am. Chem. Soc. 2022, 144, 21470–21484).
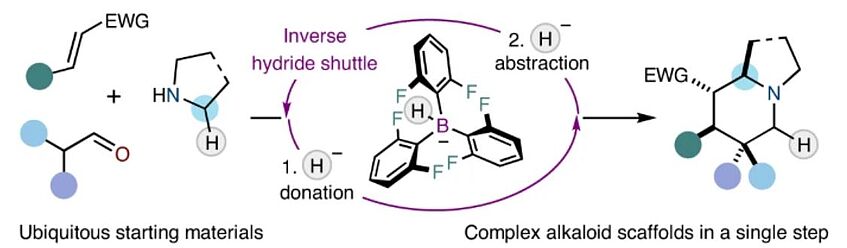
Inverse hydride shuttle catalysis enables the stereoselective one-step synthesis of complex frameworks
The rapid assembly of complex scaffolds in a single step from simple precursors identifies as an ideal reaction in terms of efficiency and sustainability. Indeed, the direct single-step synthesis of complex alkaloid frameworks remains an unresolved problem at the heart of organic chemistry in spite of the tremendous progress of the discipline. Herein, we present a broad strategy in which dynamically assembled ternary complexes are converted into valuable azabicyclic scaffolds based on the concept of inverse hydride shuttle catalysis. The ternary complexes are readily constructed in situ from three simple precursors and enable a highly modular installation of various substitution patterns. Upon subjection to a unique dual-catalytic system, the transient intermediates undergo an unusual hydride shuttle process that is initiated by a hydride donation event. Furthermore, we show that, in combination with asymmetric organocatalysis, the product alkaloid frameworks are obtained in excellent optical purity (Nat. Chem. 2022, 14, 1306–1310).
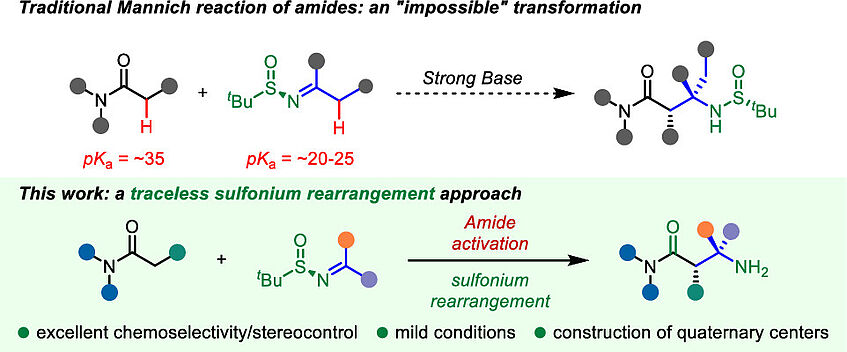
Deployment of Sulfinimines in Charge-Accelerated Sulfonium Rearrangement Enables a Surrogate Asymmetric Mannich Reaction
β-Amino acid derivatives are key structural elements in synthetic and biological chemistry. Despite being a hallmark method for their preparation, the direct Mannich reaction encounters significant challenges when carboxylic acid derivatives are employed. Indeed, not only is chemoselective enolate formation a pitfall (particularly with carboxamides), but most importantly the inability to reliably access α-tertiary amines through an enolate/ketimine coupling is an unsolved problem of this century-old reaction. Herein, we report a strategy enabling the first direct coupling of carboxamides with ketimines for the diastereo- and enantioselective synthesis of β-amino amides. This conceptually novel approach hinges on the innovative deployment of enantiopure sulfinimines in sulfonium rearrangements, and at once solves the problems of chemoselectivity, reactivity, and (relative and absolute) stereoselectivity of the Mannich process. In-depth computational studies explain the observed, unexpected (dia)stereoselectivity and showcase the key role of intramolecular interactions, including London dispersion, for the accurate description of the reaction mechanism (J. Am. Chem. Soc. 2022, 144, 13044–13049).
Direct Stereodivergent Olefination of Carbonyl Compounds with Sulfur Ylides
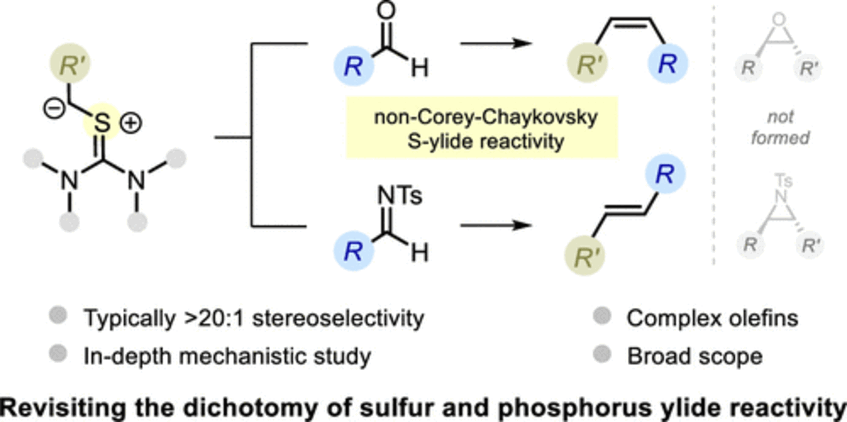
Direct Stereodivergent Olefination of Carbonyl Compounds with Sulfur Ylides
The reactivity of phosphorus and sulfur ylides toward carbonyl compounds constitutes a well-known dichotomy that is a common educational device in organic chemistry─the former gives olefins, while the latter gives epoxides. Herein, we report a stereodivergent carbonyl olefination that challenges this dichotomy, showcasing thiouronium ylides as valuable olefination reagents. With this method, aldehydes are converted to Z-alkenes with high stereoselectivity and broad substrate scope, while N-tosylimines provide a similarly proficient entry to E-alkenes. In-depth computational and experimental studies clarified the mechanistic details of this unusual reactivity (J. Am. Chem. Soc. 2022, 144, 12536–12543).
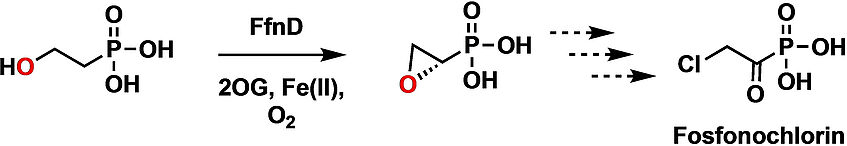
Biosynthesis of the Fungal Organophosphonate Fosfonochlorin Involves an Iron(II) and 2-(Oxo)glutarate Dependent Oxacyclase
The fungal metabolite Fosfonochlorin features a chloroacetyl moiety that is unusual within known phosphonate natural product biochemistry. Putative biosynthetic genes encoding Fosfonochlorin in Fusarium and Talaromyces spp. were investigated through reactions of encoded enzymes with synthetic substrates and isotope labelling studies. We show that the early biosynthetic steps for Fosfonochlorin involve the reduction of phosphonoacetaldehyde to form 2-hydroxyethylphosphonic acid, followed by oxidative intramolecular cyclization of the resulting alcohol to form (S)-epoxyethylphosphonic acid. The latter reaction is catalyzed by FfnD, a rare example of a non-heme iron/2-(oxo)glutarate dependent oxacyclase. In contrast, FfnD behaves as a more typical oxygenase with ethylphosphonic acid, producing (S)-1-hydroxyethylphosphonic acid. FfnD thus represents a new example of a ferryl generating enzyme that can suppress the typical oxygen rebound reaction that follows abstraction of a substrate hydrogen by a ferryl oxygen, thereby directing the substrate radical towards a fate other than hydroxylation (ChemBioChem 2022, 23, e202100352).
As Similar As Possible, As Different As Necessary — On-Site Laboratory Teaching during the COVID-19 Pandemic
As Similar As Possible, As Different As Necessary — On-Site Laboratory Teaching during the COVID-19 Pandemic
Most of the available information on studying under the challenging conditions brought about by the COVID-19 pandemic emphasizes a variety of aspects on how to digitalize the whole teaching process. Thus, several useful and potentially game-changing strategies have been reported recently. In contrast to the digitalization of teaching, in this article, we focus on the reverse process: transitioning back to offline teaching, which is unavoidable especially for the acquisition of practical skills during chemistry studies. In this work, we describe our own experience acquired during the Organic Chemistry practical course at the University of Vienna, which was held in June 2020 and onwards. The article contains descriptions of precautions and measures that were taken, additional materials, and necessary changes made in order to safely continue on-site course teaching. We anticipate that this set of precautions can be used in an adapted fashion for any type of laboratory course. Further, we offer a critical analysis of students’ and instructors’ opinions concerning the changes and well-being during the course. Those opinions were collected via a detailed survey. From our experience, with careful planning and responsible behavior, a return to on-site education is possible and warmly welcomed by all involved participants. The detailed description of our course may also be useful for those who need to start a new organic laboratory course or want to improve an existing one (J. Chem. Educ. 2021, 98, 3143–3152).
peri-Xanthenoxanthene (PXX): a Versatile Organic Photocatalyst in Organic Synthesis
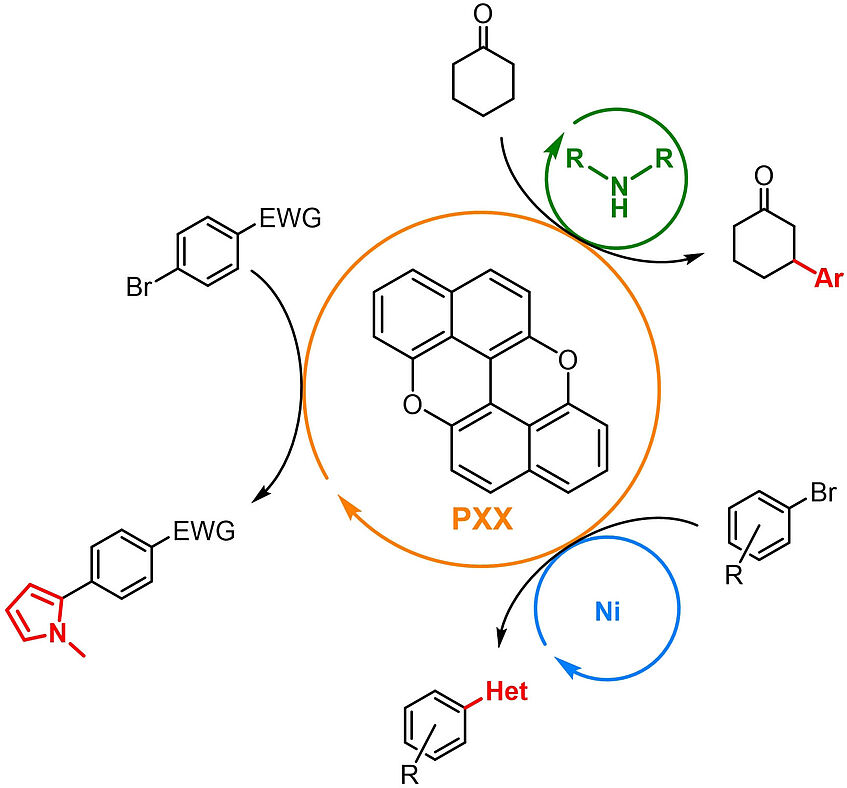
peri-Xanthenoxanthene (PXX): a Versatile Organic Photocatalyst in Organic Synthesis
Recent years have witnessed a continuous development of photocatalysts to satisfy the growing demand of photophysical and redox properties in photoredox catalysis, with complex structures or alternative strategies devised to access highly reducing or oxidising systems. We report herein the use of peri-xanthenoxanthene (PXX), a simple and inexpensive dye, as an efficient photocatalyst. Its highly reducing excited state allows activation of a wide range of substrates, thus triggering useful radical reactions. Benchmark transformations such as the addition of organic radicals, generated by photoreduction of organic halides, to radical traps are initially demonstrated. More complex dual catalytic manifolds are also shown to be accessible: the β-arylation of cyclic ketones is successful when using a secondary amine as organocatalyst, while cross-coupling reactions of aryl halides with amines and thiols are obtained when using a Ni co-catalyst. Application to the efficient two-step synthesis of the expensive fluoro-tetrahydro-1H-pyrido[4,3-b]indole, a crucial synthetic intermediate for the investigational drug setipiprant, has been also demonstrated (Adv. Synth. Catal. 2021, 363, 4740–4753).
Direct Synthesis of Enamides via Electrophilic Activation of Amides
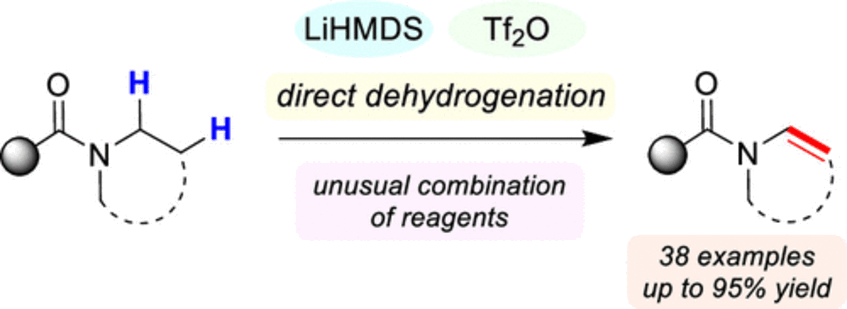
Direct Synthesis of Enamides via Electrophilic Activation of Amides
A novel, one-step N-dehydrogenation of amides to enamides is reported. This reaction employs the unlikely combination of LiHMDS and triflic anhydride, which serves as both the electrophilic activator and the oxidant, and is characterized by its simple setup and broad substrate scope. The synthetic utility of the formed enamides was readily demonstrated in a range of downstream transformations (J. Am. Chem. Soc. 2021, 143, 10524–10529).
Nuclear Magnetic Relaxation Mapping of Spin Relaxation in Electrically Stressed Glycerol
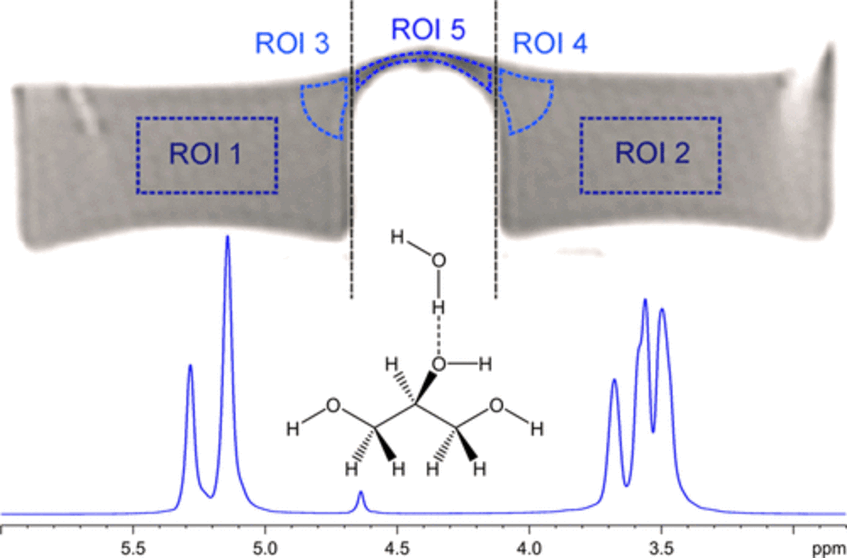
Nuclear Magnetic Relaxation Mapping of Spin Relaxation in Electrically Stressed Glycerol
This work discusses nuclear magnetic relaxation effects in glycerol subject to a strong electric field. The methods used are 1.5 T magnetic resonance imaging (MRI), referenced by 9.4 T nuclear magnetic resonance (NMR). While MRI allows a glycerol probe to be sampled with a high voltage (HV) of 16 kV applied to the probe, NMR provides precise molecular data from the sample, but the sample cannot be tested under HV. Using MRI, the recording of magnetic relaxation times was possible while HV was applied to the glycerol. NMR spectroscopy was used to confirm that MRI provides a reasonably accurate estimation of temperature. The applied HV was observed to have a negligible effect on the spin–lattice relaxation time T1, which represents the energy release to the thermal bath or system enthalpy. In contrast to that, the spin–spin relaxation time T2, which does represent the local entropy of the system, shows a lower response to temperature while the liquid is electrically stressed. These observations point toward a proton population in electrically stressed glycerol that is more mobile than that found in the bulk, an observation that is in agreement with previously published results for water (ACS Omega 2020, 5, 22057–22070).
Combining high-resolution scanning tunnelling microscopy and first-principles simulations to identify halogen bonding
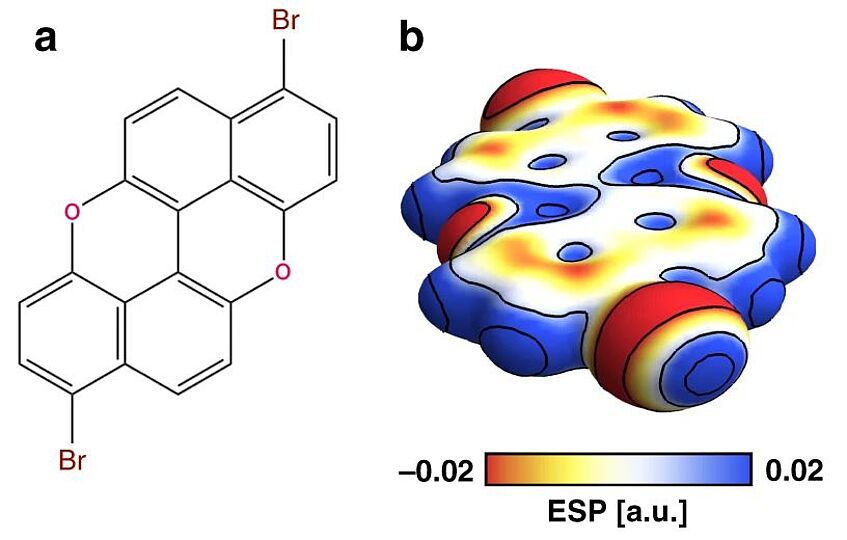
Combining high-resolution scanning tunnelling microscopy and first-principles simulations to identify halogen bonding
Scanning tunnelling microscopy (STM) is commonly used to identify on-surface molecular self-assembled structures. However, its limited ability to reveal only the overall shape of molecules and their relative positions is not always enough to fully solve a supramolecular structure. Here, we analyse the assembly of a brominated polycyclic aromatic molecule on Au(111) and demonstrate that standard STM measurements cannot conclusively establish the nature of the intermolecular interactions. By performing high-resolution STM with a CO-functionalised tip, we clearly identify the location of rings and halogen atoms, determining that halogen bonding governs the assemblies. This is supported by density functional theory calculations that predict a stronger interaction energy for halogen rather than hydrogen bonding and by an electron density topology analysis that identifies characteristic features of halogen bonding. A similar approach should be able to solve many complex 2D supramolecular structures, and we predict its increasing use in molecular nanoscience at surfaces (Nat. Commun. 2020, 11, 2103).
An α-Cyclopropanation of Carbonyl Derivatives by Oxidative Umpolung
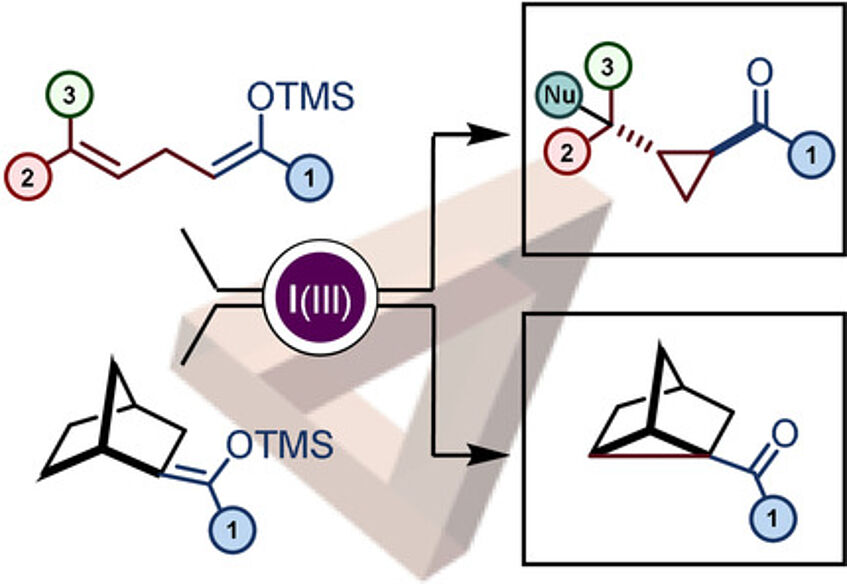
An α-Cyclopropanation of Carbonyl Derivatives by Oxidative Umpolung
The reactivity of iodine(III) reagents towards nucleophiles is often associated with umpolung and cationic mechanisms. Herein, we report a general process converting a range of ketone derivatives into α-cyclopropanated ketones by oxidative umpolung. Mechanistic investigation and careful characterization of side products revealed that the reaction follows an unexpected pathway and suggests the intermediacy of non-classical carbocations (Angew. Chem. Int. Ed. 2020, 59, 18208–18212).
Application of Relay C−H Oxidation Logic to Polyhydroxylated Oleanane Triterpenoids
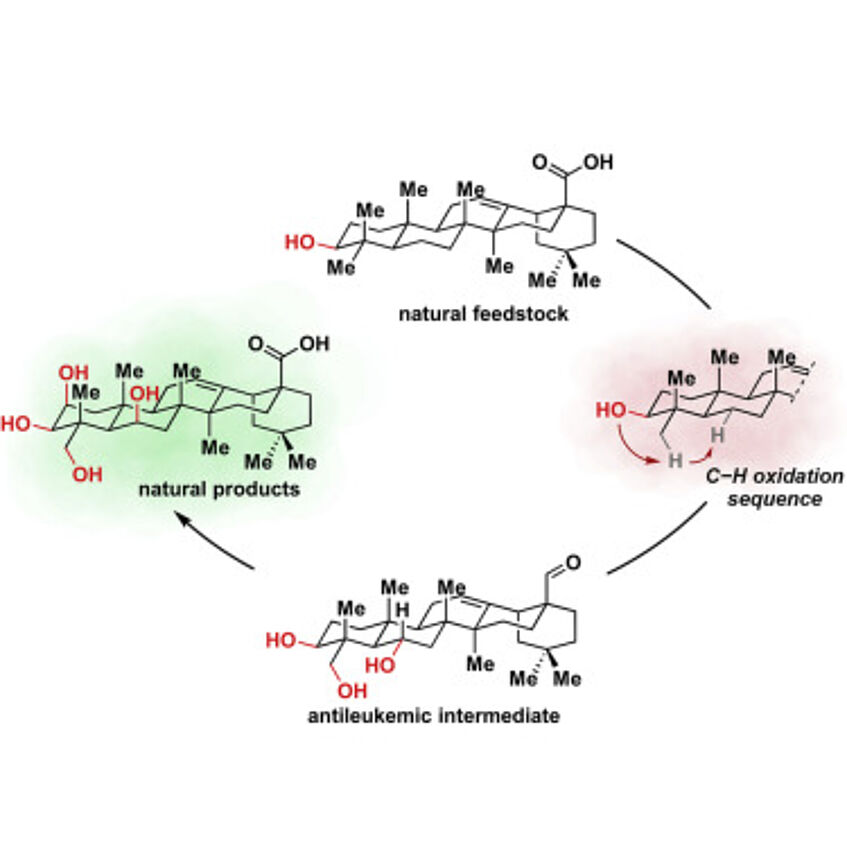
Application of Relay C−H Oxidation Logic to Polyhydroxylated Oleanane Triterpenoids
Chemical modification of readily available, scarcely oxidized natural products not only enables access to higher and more valuable congeners but can also pave the way to pharmaceutically relevant derivatives with enhanced properties. Achieving site-selective oxidation of such natural products is an endeavor often controlled by the presence of directing elements. Because of this fact, the oxidation of C−H bonds remote from such elements is either unfeasible or requires more elaborate synthetic strategies. We report the use of relay C−H oxidation as a concept to achieve the oxidation of previously inaccessible ring B in the abundant feedstock oleanolic acid. This resulted not only in the first total synthesis of a number of polyhydroxylated natural triterpenoids but also revealed an anti-leukemic intermediate. The strategy presented here could become a very general approach in the synthesis of valuable triterpenoids (Chem 2020, 6, 1183–1189).
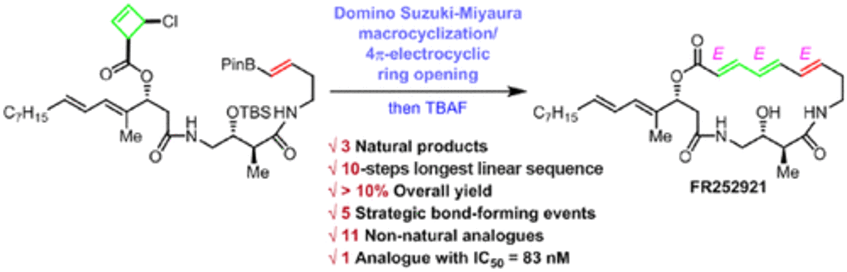
A Domino 10-Step Total Synthesis of FR252921 and Its Analogues, Complex Macrocyclic Immunosuppressants
FR252921, FR252922, and FR256523 are a family of potent macrocyclic polyene immunosuppressive agents with a novel mode of action. However, the lack of an efficient and flexible synthesis has hindered further biological studies, mostly due to the fact that the natural products appear to be kinetic isomers regarding the triene moiety. Herein, we report the development and application of an unprecedented, unique domino Suzuki–Miyaura/4π-electrocyclic ring-opening macrocyclization, resulting in a concise, unified, and stereoselective synthetic route to these complex targets in only 10 steps. This in turn enables ready access to a range of unnatural analogues, among which several compounds showed inhibition of T-lymphocyte proliferation at levels equal or superior to those of the natural products themselves (J. Am. Chem. Soc. 2019, 141, 13772–13777).
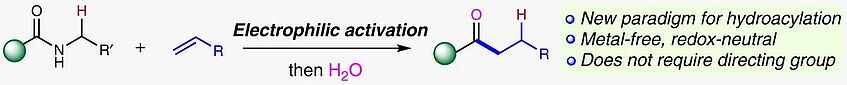
A redox-neutral synthesis of ketones by coupling of alkenes and amides
The direct synthesis of ketones via carbon–carbon bond formation represents one of the most important challenges in organic synthesis. Hydroacylation of alkenes offers perhaps the most efficient and atom-economical approach for the preparation of ketones employing carbonyl compounds and alkenes as feedstocks. State-of-the-art hydroacylation is typically achieved by a transition metal-catalysed coupling of an aldehyde and an alkene but is plagued by competing decarbonylation, requiring the installation of directing groups in the aldehyde reactant. Herein, we present a method for the hydroacylation of alkenes employing amides in a metal-free regime, proceeding by a new mechanism and offering orthogonal reactivity to the conventional, metal-catalysed alternatives (Nat. Commun. 2019, 10, 2327).
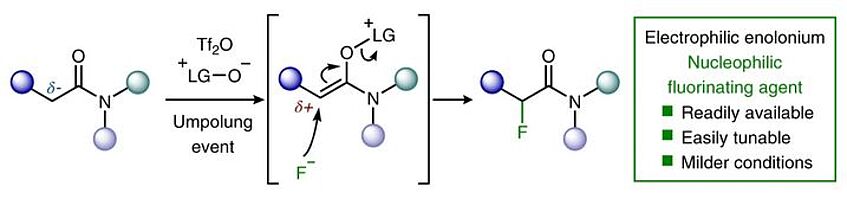
α-Fluorination of carbonyls with nucleophilic fluorine
Given the unique properties of fluorine, and the ability of fluorination to change the properties of organic molecules, there is significant interest from medicinal chemists in innovative methodologies that enable the synthesis of new fluorinated motifs. State-of-the-art syntheses of α-fluorinated carbonyl compounds invariably rely on electrophilic fluorinating agents, which can be strongly oxidizing and difficult to handle. Here we show that reversing the polarity of the enolate partner to that of an enolonium enables nucleophilic fluorinating agents to be used for direct chemoselective α-C–H-fluorination of amides. Reduction of these products enables facile access to β-fluorinated amines and the value of this methodology is shown by the easy preparation of a number of fluorinated analogues of drugs and agrochemicals. A fluorinated analogue of citalopram, a marketed antidepressant drug, is presented as an example of the preserved biological activity after fluorination (Nat. Chem. 2019, 11, 329–334).
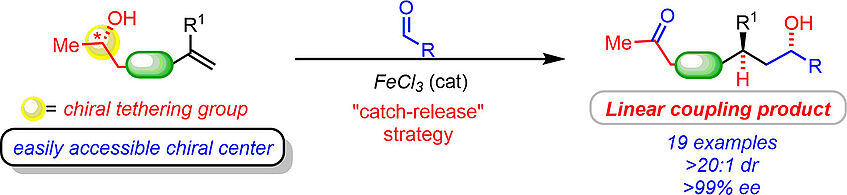
Enantioselective Redox-Neutral Coupling of Aldehydes and Alkenes by an Iron-Catalyzed “Catch–Release” Tethering Approach
The reductive coupling of aldehydes and alkenes is an emerging technology that holds the potential to reinvent carbonyl addition chemistry. However, existing enantioselective methods are limited to form “branched” products. Herein, we present a directed enantio- and diastereoselective alkylation of aldehydes with simple olefins to selectively yield linear coupling products. This is achieved by redox-neutral remote functionalization, whereby a tethering “catch–release” strategy decisively solves the key problems of reactivity and selectivity (J. Am. Chem. Soc. 2019, 141, 143–147).
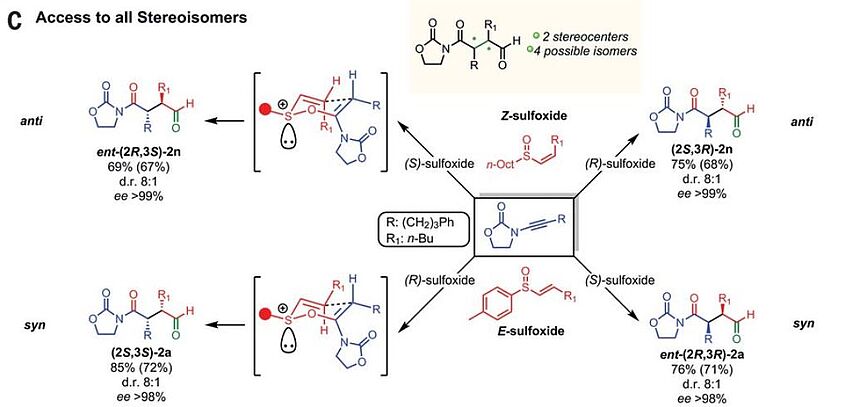
Stereodivergent synthesis of 1,4-dicarbonyls by traceless charge–accelerated sulfonium rearrangement
The chemistry of the carbonyl group is essential to modern organic synthesis. The preparation of substituted, enantioenriched 1,3- or 1,5-dicarbonyls is well developed, as their disconnection naturally follows from the intrinsic polarity of the carbonyl group. By contrast, a general enantioselective access to quaternary stereocenters in acyclic 1,4-dicarbonyl systems remains an unresolved problem, despite the tremendous importance of 2,3-substituted 1,4-dicarbonyl motifs in natural products and drug scaffolds. Here we present a broad enantioselective and stereodivergent strategy to access acyclic, polysubstituted 1,4-dicarbonyls via acid-catalyzed [3,3]-sulfonium rearrangement starting from vinyl sulfoxides and ynamides. The stereochemistry at sulfur governs the absolute sense of chiral induction, whereas the double bond geometry dictates the relative configuration of the final products (Science 2018, 361, 664–667).
Total Synthesis of Quinine

Total Synthesis of Quinine
One of the research interests of the Maulide Group is the total synthesis of natural products. Recently, the group developed a novel synthetic route to the well-known alkaloid quinine (1). Quinine, which was isolated first from the bark of the Cinchona tree in 1820, can be found in tonic water where it is responsible for the bitter taste. Furthermore, quinine is used as a potent anti-malarial drug, especially against resistant strains.
The developed route which utilizes modern catalytic C–H activation – these are reactions that permit controlled cleavage of carbon-hydrogen bonds – allows the synthesis of both quinine enantiomers – the natural (–)-quinine as well as the unnatural (+)-quinine – in a short number of steps. A slight change in the synthetic sequence, furthermore, enabled the synthesis of unprecedented C3-aryl analogues. Two of these analogues, 2a,b, have been tested against a malaria pathogen, Plasmodium berghei, in mice. While both analogues showed anti-malarial activity, the CF3-substituted 2a outperformed the natural product in both administered doses and led to a three-fold increased survival time.
All results and details can be accessed free of charge in the open access article published in Angewandte Chemie International Edition (English, Angew. Chem. Int. Ed. 2018, 57, 10737–10741.) and Angewandte Chemie (German, Angew. Chem. 2018, 130, 10897–10901.).
Highly Selective Stable Isotope Labeling of Histidine Residues by Using a Novel Precursor in E. coli-Based Overexpression Systems
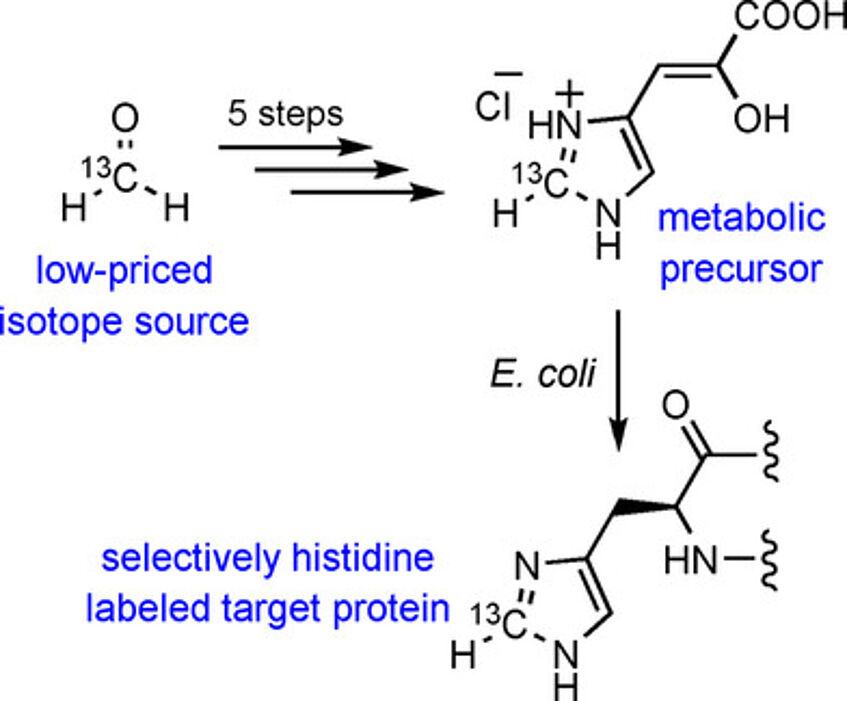
Highly Selective Stable Isotope Labeling of Histidine Residues by Using a Novel Precursor in E. coli-Based Overexpression Systems
The importance of NMR spectroscopy in unraveling the structural and dynamic properties of proteins is ever-expanding owing to progress in experimental techniques, hardware development, and novel labeling approaches. Multiple sophisticated methods of aliphatic residue labeling can be found in the literature, whereas the selective incorporation of NMR active isotopes into other amino acids still holds the potential for improvement. In order to close this methodological gap, we present a novel metabolic precursor for cell-based protein overexpression to assemble 13C/2H isotope patterns in the peptide backbone, as well as in side chain positions of a mechanistically distinguished histidine residue (ChemBioChem 2017, 18, 1487–1491).
Hydroxamic Acids as Chemoselective (ortho-Amino)arylation Reagents via Sigmatropic Rearrangement
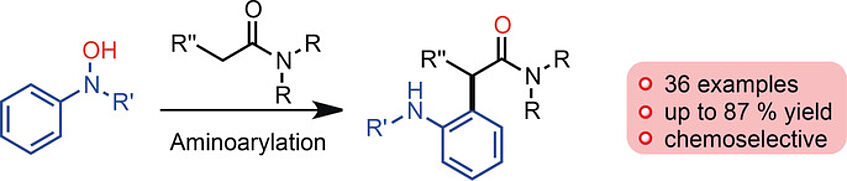
Hydroxamic Acids as Chemoselective (ortho-Amino)arylation Reagents via Sigmatropic Rearrangement
The use of readily available hydroxamic acids as reagents for the chemoselective (ortho-amino)arylation of amides is described. This reaction proceeds under metal-free, mild conditions, displays a very broad scope, and constitutes a direct approach for the metal-free attachment of aniline residues to carbonyl derivatives (Angew. Chem. Int. Ed. 2017, 56, 10938–10941).
Chemoselective Intermolecular Cross-Enolate-Type Coupling of Amides

Chemoselective Intermolecular Cross-Enolate-Type Coupling of Amides
A new approach for the synthesis of 1,4-dicarbonyl compounds is reported. Chemoselective activation of amide carbonyl functionality and subsequent umpolung viaN-oxide addition generates an electrophilic enolonium species that can be coupled with a wide range of nucleophilic enolates. The method conveys broad functional group tolerance on both components, does not suffer from formation of homocoupling byproducts and avoids the use of transition metal catalysts (J. Am. Chem. Soc. 2017, 139, 45, 16040–16043).
New Applications of Selective Isotopic Labeling of Proteins
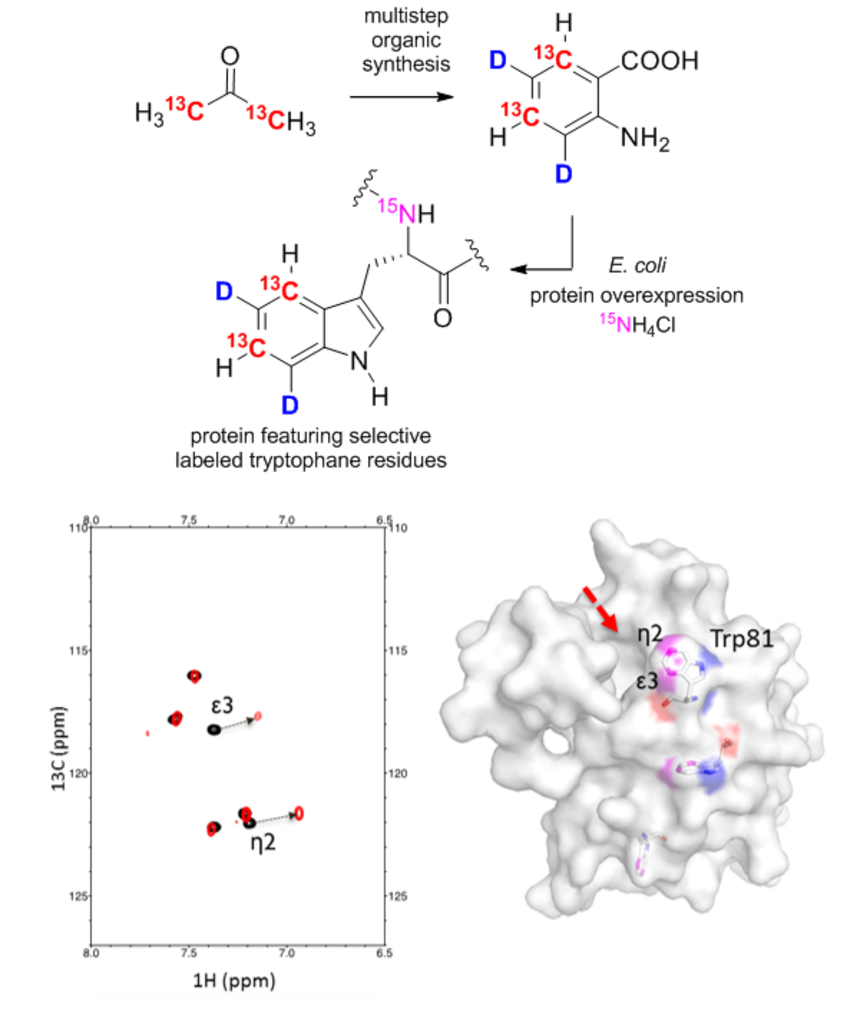
Top: Synthesis of isotopically labeled anthranilic acid from 13C2-acetone and expression of a selectively Trp-labeled protein. Bottom: Studies of the ligand binding of a protein using NMR spectroscopy.
New Applications of Selective Isotopic Labeling of Proteins
In recent years, the group led by Roman Lichtenecker has developed a range of syntheses to make various isotopically labeled amino acid precursors accessible. These molecules possess specific 13C, 15N, and 2D patterns and are used for the selective isotopic labeling of proteins. In this process, the compounds can be added to the growth media of recombinant E. coli strains that overexpress a specific target protein. The isotopically labeled precursors are then metabolically converted in vivo by the microorganisms into the desired amino acids. This approach opens remarkable new possibilities for elucidating protein structures, dynamic properties, or ligand binding using NMR spectroscopy, particularly for the aromatic amino acids phenylalanine, tyrosine, tryptophan, and histidine.In collaboration with the group led by Prof. Robert Konrat at the Department of Structural Biology and Computational Biology, new examples of applications for these compounds have been demonstrated. The results have been published in the Journal of Biomolecular NMR and the article is freely accessible as an open access publication (J. Biomol. NMR 2018, 71, 129–140).